



Abstract
Having a production line for microrobots is fundamental to their widespread application in many areas. Thanks to their flexibility, active colloidal molecules with dynamic function are a promising model for microrobots. So far, the production of colloidal molecules requires a great deal of engineering and ad hoc processes. Here, we first describe a method for the continuous production of dimers and trimers formed by non-polar spherical active colloidal particles. We use external fields to both guide the monomers and rotate the colloidal molecules as to obtain dimers and trimers with very high yield and highly localized in space. We also show how fields and rigid walls can be used to direct small colloidal molecules to specific locations. Finally, we study the possible colloidal molecules that can be created with these building blocks and propose a general mechanism for the directed assembly of colloidal molecules with dynamic function.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Active colloidal particles are one of the promising candidates to serve as microrobots in possible biomedical solutions a la 'Fantastic Voyage' film [1]: small machines that can travel and fulfill various complex tasks inside the body. Active colloids can self-propel by several mechanisms: bubble ejection [2, 3], different kind of phoresis [4], and beating or twisting like flagella [5–8].
There are several methods for the production of colloidal structures in use, for example, top-down approaches using DNA adhesion [9–11], electric or magnetic field manipulation [8, 12, 13], random formation aided by light [14] or spontaneous clustering [15], dependent on the way energy is injected into the system [16] or by geometrical considerations [17]. By these means, several structures have been found: swimmers, rotators, helixes, chains and all kind of compact clusters. These methods work well but there is no clear way of how to generalize these approaches to produce particular isomers. In this paper we show how it could be possible to design factories that direct the self-assembly of colloidal particles and produce, with a very high yield, isomers that are otherwise almost impossible to form by chance [18]. Furthermore, by a careful selection of the design parameters, we can tailor the resulting isomer.
In a previous work we have shown how chains of non-polar active colloids can spontaneously oscillate and behave as cilia or flagella, but we did not present a method to assemble those structures [19]. Despite the fact that several methods for the design of complex flagella-like structures have been developed, for example clusters of particles with elastic bonds [8, 12], or chains sustained by magnetic fields [20], there is no general procedure to create the type of chains that we have previously studied. The aim of this paper is to describe one such method.
The motivation for a colloidal factory are the recent studies on how active colloidal particles can be guided by topographical pathways [21], external light gradients [22] or 'fuel' concentration [23, 24]. These experiments were realized with Janus particles; we show here that the same applies to self-assembled colloidal molecules that are held together only by mutual soft interactions as the phoretic forces. In particular, we show that dimers and trimers are stable enough to be guided by external traps and move almost in the line defined by an effective potential's minimum.
The fact that these modular microswimmers [25, 26] are soft enough to reassemble upon collision, but stiff enough to be guided by walls and chemical channels, allows us to simulate different components of a colloidal factory: a production zone for dimers and trimers; channels where those colloids can be guided; walls that can be used to contain and deviate the colloids; and finally, the directed collision of small molecules as to form oscillators with a high yield.
The article is organized as follows. First, we present the model system and its numerical implementation. Second, we show how, by using external fields and a certain geometry, we can transport monomers to a reaction zone, where they produce dimers and trimers with high yield. Third, we study how these dimers and trimers can be guided by external potentials and walls. Finally we study how, by guided collisions, trimers can form more complex structures with a clear preference in the formed isomer depending on the parameters of the collision and the chemistry of the particles.
2. The model
2.1. Equations of motion
For the construction of the cilia- and flagella-like structures described in [19], we considered a simple model of active particles, which do not self-propel on their own, but rather activity emerges as a result of interactions [27]. In presence of concentration inhomogeneities, colloids experience a phoretic drift which is well described by a velocity proportional to the concentration gradient of a relevant solute, multiplied by a mobility coefficient μ,  [28]. This concentration field can be generated by external sources and sinks
[28]. This concentration field can be generated by external sources and sinks  , but also it can be produced by catalytic reactions on the particles surfaces themselves. Active colloidal particles, for example, gold or platinum particles in a water peroxide solution, generate or consume solutes at their surfaces, which later diffuse on the fluid. Janus particles self-propel by this mechanism [29, 30]. Here, we consider isotropic colloids, each one made of a pure material (e.g. gold or platinum). For micron sized particles, with speeds of few microns per second, the solute Péclet number is small; hence, we use the approximation where the solute concentration profiles are completely enslaved to the instantaneous positions of the colloids
, but also it can be produced by catalytic reactions on the particles surfaces themselves. Active colloidal particles, for example, gold or platinum particles in a water peroxide solution, generate or consume solutes at their surfaces, which later diffuse on the fluid. Janus particles self-propel by this mechanism [29, 30]. Here, we consider isotropic colloids, each one made of a pure material (e.g. gold or platinum). For micron sized particles, with speeds of few microns per second, the solute Péclet number is small; hence, we use the approximation where the solute concentration profiles are completely enslaved to the instantaneous positions of the colloids  and the production rates at their surfaces αi. The case of finite Péclet number gives rise to interesting fenomena as it has been reported in [31]. In the far field approximation, the interaction between two colloids in three dimensions decays as the distance squared and the resulting diffusiophoretic velocity of the colloid i interacting with colloid k is
and the production rates at their surfaces αi. The case of finite Péclet number gives rise to interesting fenomena as it has been reported in [31]. In the far field approximation, the interaction between two colloids in three dimensions decays as the distance squared and the resulting diffusiophoretic velocity of the colloid i interacting with colloid k is  , where
, where  is the vector that joins the centers of the particles and U0 depends on their radii and the diffusion coefficient of solutes [27]. Notably, the interaction between two dissimilar particles is not symmetric, violating the action-reaction principle. This is possible by the presence of the supporting fluid that takes away or provides momentum, and it is the source of activity in this model. Besides this interaction, colloidal particles display steric repulsion, which we model as a soft-sphere potential. The equations of motion for overdamped particles in presence of Brownian noise read
is the vector that joins the centers of the particles and U0 depends on their radii and the diffusion coefficient of solutes [27]. Notably, the interaction between two dissimilar particles is not symmetric, violating the action-reaction principle. This is possible by the presence of the supporting fluid that takes away or provides momentum, and it is the source of activity in this model. Besides this interaction, colloidal particles display steric repulsion, which we model as a soft-sphere potential. The equations of motion for overdamped particles in presence of Brownian noise read
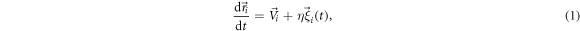
with
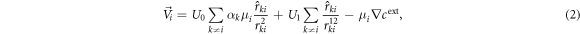
where U1 measures the intensity of the repulsive force and η is the noise intensity, which gives rise to a bare diffusion coefficient for monomers, D = η2/2. Here,  is a vectorial white noise of zero mean and correlation
is a vectorial white noise of zero mean and correlation  , with i, k labeling particles and a, b indicating the Cartesian coordinates. Although the pair interaction has been derived in three spatial dimensions, as colloidal particles normally sediment, their motion will be restricted to two dimensions.
, with i, k labeling particles and a, b indicating the Cartesian coordinates. Although the pair interaction has been derived in three spatial dimensions, as colloidal particles normally sediment, their motion will be restricted to two dimensions.
For a binary mixture (types A and B) with the signs of the phoretic charges chosen such that (αA, μA) = (+, +) and (αB, μB) = (−, −), equal particles repel and dissimilar particles attract. At low concentration of colloidal particles, this results in the formation of clusters or colloidal molecules, where A and B particles alternate. The smallest of these are dimers AB and trimers AB2. Depending on the values of the charges, colloidal molecules can be stable in different isomer configurations. For example, the trimer can adopt a linear configuration, but also, for a wide range of parameters, it adopts a polar triangular shape. The dimer is also polar and both present self-propulsion as a result of the violation of the action-reaction principle [27].
It is possible to absorb the dimensions of α and μ into U0, allowing us to chose αB = −1 and μB = −1, leaving αA and μA (both positive) as free dimensionless variables. Also, time and space units can be chosen such that U0 = U1 = 1. Alternatively, keeping dimensional forms for U0 and U1, the length, time, and velocity units are  ,
,  , and
, and  , respectively. The minimal distance between particles that attract each other is of order l0, therefore fixing the hard-core diameter. For comparison, in the experiments described in [25], l0 ≈ 10 μm and v0 ≈ 1 μm s−1, resulting in t0 ≈ 1 s. Similar molecules have been found with different choices of the mobilities' signs [25, 26, 32] but we are not aware that the oscillating chain can be formed like that, so we restrict ourselves to the present case.
, respectively. The minimal distance between particles that attract each other is of order l0, therefore fixing the hard-core diameter. For comparison, in the experiments described in [25], l0 ≈ 10 μm and v0 ≈ 1 μm s−1, resulting in t0 ≈ 1 s. Similar molecules have been found with different choices of the mobilities' signs [25, 26, 32] but we are not aware that the oscillating chain can be formed like that, so we restrict ourselves to the present case.
2.2. Effect of the external concentration field
Strong inhomogeneities on the field can break colloidal molecules by tidal effects (and will be studied in detail in section 4), but at moderate values, colloidal molecules will behave as solid bodies and the field will move and rotate them [22]. For isotropic clusters composed of N particles, the external field produces a net velocity. It is proportional to the concentration gradient evaluated at the geometric center of the cluster,  , and the cluster's average mobility,
, and the cluster's average mobility,  , plus corrections that depend on the second derivatives of cext. For anisotropic clusters, besides this velocity, there is an induced angular velocity
, plus corrections that depend on the second derivatives of cext. For anisotropic clusters, besides this velocity, there is an induced angular velocity  , which is computed as follows. Defining the relative coordinates
, which is computed as follows. Defining the relative coordinates  , it is direct that
, it is direct that  where the gradient is evaluated at
where the gradient is evaluated at  by neglecting contributions from higher derivatives. Assuming that the cluster does not deform or break thanks to the balance of the phoretic and repulsive forces, the solid body rotation implies also,
by neglecting contributions from higher derivatives. Assuming that the cluster does not deform or break thanks to the balance of the phoretic and repulsive forces, the solid body rotation implies also,  . Equating both expressions and using that in two dimensions
. Equating both expressions and using that in two dimensions  and
and  are perpendicualr, gives
are perpendicualr, gives  , where
, where  is the dipolar moment of the cluster. How fast these molecules align and what trajectory they follow depend on the geometry of the molecule; thus, for example, dimers turn much faster than trimers in an external field (see figure 1). These formulas are for illustration purposes only, we actually integrate the particle-particle interaction in an external field for field strengths small enough as to not break any of the formed molecules.
is the dipolar moment of the cluster. How fast these molecules align and what trajectory they follow depend on the geometry of the molecule; thus, for example, dimers turn much faster than trimers in an external field (see figure 1). These formulas are for illustration purposes only, we actually integrate the particle-particle interaction in an external field for field strengths small enough as to not break any of the formed molecules.

Figure 1. Trajectories of small colloidal molecules in the presence of a constant concentration gradient ∇cext, shown as gray arrows. Monomers simply follow the gradient in a direction that depends on the sign of the mobility: against or along the gradient for A (red) and B (cyan) particles, respectively. Self-propelled molecules (dimers and trimers) are aligned with the gradient, with different speeds: the smaller the molecule the fastest is the alignment. Clusters were simulated in isolation for 150 time units and they are put together in the figure for illustration purposes only. In all cases, the values of the parameters are αA = 1.5, μA = 0.5, αB = −1, μB = −1, η = 0, and  .
.
Download figure:
Standard image High-resolution image3. Production of dimers and trimers
3.1. System geometry and external field
For the efficient production of dimers and trimers, we take an external concentration field with a geometry that focuses particles in a small reaction zone. This field is assumed to be produced by sources and sinks far from the reaction zone. For this, it is chosen to be a solution of the stationary diffusion equation, built using the conformal mapping technique. There are two impenetrable walls that leave a small opening of width 2b0, where monomers and colloidal molecules can interact (see figure 2). We take
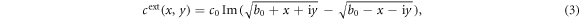
where Im is the imaginary part and c0 measures the field intensity. To avoid colloids to reach and cross the walls, an additional repulsive potential is added

where P0 = r0 = 1, such that the induced velocity on each colloid is  (equal for all particles).
(equal for all particles).

Figure 2. Scheme of the configuration used to produce dimers and trimers. A and B particles inserted at the red and cyan regions, respectively, will travel either moving against (A) or along (B) the concentration gradient, whose flow lines are shown in gray. Particles are taken out from the simulation when crossing the black outer circle of radius 50. Impenetrable walls (red) are added on the x axis, with an opening region of width 2b0 = 8, where particles can interact.
Download figure:
Standard image High-resolution imageIf A and B monomers are inserted at the rectangular regions in figure 2, located at opposite sides of the reaction zone, they will move along the field lines and meet near the origin where they can interact and assembly into colloidal molecules. Since we aim for particles to arrive at the origin at roughly the same time, we need to put particles at different locations, for they have different mobilities. Each particle is injected with a uniform probability in a rectangle of size 2 × 6 in the x and y direction, respectively. This mimics particles that are let to sediment from the third dimension into these regions in microfluidic devices. The insert location is such that the particles tend to meet at the center of the system. The distance between the two insertion points permits the particles to first form a dimer, then change their direction of movement, and after that collide with another monomer to form a trimer. See supplementary video 1 available online at stacks.iop.org/NJP/21/033041/mmedia for an example.
The results presented here are in the diluted-injection regime, that is, there are only a few monomers in the volume at any time, and the times between insertions are comparable to the time it takes a monomer to travel the system due to the effect of the external field only. In this way, we ensure that the majority of reactions in the system are monomer to monomer, and monomer to dimer, and colloidal molecules are always isolated so no larger reaction occurs.
Since we aim to simulate the continuous production of colloidal molecules, the total number of particles in our simulation is not conserved: as we inject particles their number increases, and for the sake of simplicity, we remove them when they are far enough from the production zone. The insertion of particles happens at fixed time intervals Δti, where i = A, B indicates the species of the particles. Particle removal occurs when the first particle of a cluster crosses the circular boundary of radius R = 50. Note that dimers and trimers leave the system by self-propulsion whereas the monomers move passively following the external field lines as can be seen in figure 1.
Thanks to the external field, the monomers do not stick around the injection area and collapse into large amorphous clusters of particles. With the presence of the external field the unused monomers will leave the reaction area, facilitating the organized formation of dimers or trimers alone.
3.2. Results
In the regime we are studying, the most important control parameters are the fluxes of incoming particles, Ji = 1/Δti, which determine what molecules are formed. Figure 3 presents the fraction of monomers, dimers and trimers that exit the system as a function of the flux ratio JB/JA. Simulations were performed with η = 0. Note that for the zero noise case, corresponding to a vanishing temperature, if the system were not driven but let to relax freely, it would have collapsed to a single large cluster [27]. Here, the driving imposed by the external field allows us to steadily produce small colloidal molecules.

Figure 3. Fraction of clusters made of one, two or three colloids that leave the simulation domain for different flux ratios (the flux for B particles is fixed at JB = 1/150). We ran each simulation until 100 A particles are inserted for each JB/JA value. The other parameters are fixed to b0 = 2, αA = 1.7, μA = 0.4, η = 0, and c0 = 0.525. Lines, to help visualization, are obtained by averaging neighboring points.
Download figure:
Standard image High-resolution imageWhen the fluxes of A and B particles are the same, the system forms mostly dimers. By increasing the flux of B particles, the chances to form trimers increase. One would expect that a flux of B particles twice larger will create mostly trimers, but this is actually not the case. For JB = 2JA, many B colloids escape as monomers, without reacting to form colloidal molecules. The maximum production of trimers occurs for JB ≈ 1.7JA, in which case, 60% of the collected clusters are trimers.
The angular distribution of the produced colloidal molecules is shown in figure 4. Notably, dimers and timers are well focused and separated from monomers. A further separation between dimers and trimers could be arranged adding a perpendicular component to the external field. To have an idea of how a perpendicular field separates dimers of trimers is enough to look at figure 1 and how dimmers and trimers that are very close at one point during their trajectory (lower part of the figure) are separated thanks to the external field. By separating these small molecules one can use them to produce larger functional structures, as shown in the next sections.

Figure 4. Histograms of exit angles for monomers, dimers and trimers for the simulation parameters of figure 3. Data is collected for fluxes in the range 1.5 ≤ JB/JA ≤ 2, where the maximum production of trimers take place. θ is the usual polar angle, measured counter-clockwise from the x axis. The monomers (in this case B particles) leave from the bottom of the system (θ ≈ −π/2), whereas dimers and trimers leave from the top. The trimers present a smooth distribution around at θ ≈ π/2, whereas the dimers have a bi-modal distribution to each side of the vertical with a more prominent peak towards the right side of the system. The peaks at 0 and −π are particles that left the system guided by the walls. The total number of counts is 931.
Download figure:
Standard image High-resolution imageThe results presented in this section are qualitatively robust for a wide range of parameters. We have changed the insert location, field strength, opening between the walls and chemistry of the particles.
The values b = 2 and c0 = 0.525 used for the simulation shown in figures 3 and 4 were chosen after an exploration of different possibilities to produce dimers and trimers with high yield and angular focalization for the specified values of the phoretic charges. Changing b0 varies the production fractions, but no systematic tendency was detected. However, increasing b0 widens the angular distribution, losing the focalization shown in figure 4. Increasing c0 reduces the production of trimers because the monomers are driven too fast and escape the reaction zone before meeting another monomer, while decreasing its value leads to an accumulation of monomers in the reaction zone, resulting in the formation of large clusters. This problem is solved if the fluxes are reduced in proportion.
Finally, other values of the phoretic changes αA and μA in the range of parameters where dimers are self-propelled and trimers are stable (see [27]), have the same qualitatively results. The values of the optimal field strengths, opening size and flux ratios change accordingly. The addition of moderate noise does not change this picture qualitatively since monomers are carried by the external field towards the reaction area.
4. Guiding dimers and trimers
Having demonstrated the ability to create small colloidal molecules, we now show that these molecules remain stable in different external potentials and when they interact with walls.
4.1. Motion in guiding fields
In [32], it was shown that Janus particles can be guided in chemical landscapes. Here, we show that the same is possible in self-assembled colloids, with the precaution that the chemical gradient should not break the colloidal molecules apart. Consider an AB dimer moving in a external concentration field cext(x, y) = λy2/2. We assume for the moment that λ is small enough not to break the dimer and, a posteriori, we determine the maximum allowed value. The velocity of each colloid, which results from the mutual interaction and with the external field is
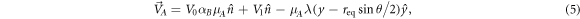

where  is the unit vector pointing from B to A, y is the coordinate of the center of mass,
is the unit vector pointing from B to A, y is the coordinate of the center of mass,  and
and  , with req the equilibrium distance of the two colloids (of order 1). We assume as in [19, 27] that the radial distance is a hard degree of freedom, which does does not vary even in presence of an external field. Imposing that the relative velocity
, with req the equilibrium distance of the two colloids (of order 1). We assume as in [19, 27] that the radial distance is a hard degree of freedom, which does does not vary even in presence of an external field. Imposing that the relative velocity  vanishes in the
vanishes in the  direction, gives
direction, gives  , where for simplicity we assumed that req ≪ y. Then, analyzing the velocity of the center of mass and the angular component of the relative velocity gives the coupled equations
, where for simplicity we assumed that req ≪ y. Then, analyzing the velocity of the center of mass and the angular component of the relative velocity gives the coupled equations
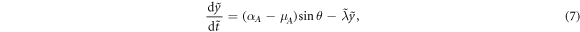
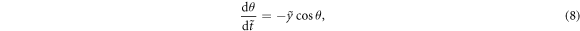
where  and
and  are rescaled variables and
are rescaled variables and ![$\tilde{\lambda }=\sqrt{\lambda {r}_{\mathrm{eq}}/[2{V}_{0}({\mu }_{A}+1)]}({\mu }_{A}-1)$](https://content.cld.iop.org/journals/1367-2630/21/3/033041/revision2/njpab0cc1ieqn32.gif) is a control parameter. For αA − μA > 0 and μA > 1 (
is a control parameter. For αA − μA > 0 and μA > 1 ( ), the configuration with the dimer moving along the valley of the concentration field (y = and θ = 0, π) is a stable equilibrium, which is achieved via damped oscillations, as shown in the phase portrait of figure 5. When αA − μA > 0 and μA < 1 this configuration is unstable and the dimer escapes from the valley. When αA − μA < 0 the referred configuration becomes a hyperbolic point and the resultant dynamics is not interesting for focusing dimers. Besides the stability, it must be imposed that the tidal forces do not break the dimer. This is guaranteed by requiring that V1 > 0, which gives
), the configuration with the dimer moving along the valley of the concentration field (y = and θ = 0, π) is a stable equilibrium, which is achieved via damped oscillations, as shown in the phase portrait of figure 5. When αA − μA > 0 and μA < 1 this configuration is unstable and the dimer escapes from the valley. When αA − μA < 0 the referred configuration becomes a hyperbolic point and the resultant dynamics is not interesting for focusing dimers. Besides the stability, it must be imposed that the tidal forces do not break the dimer. This is guaranteed by requiring that V1 > 0, which gives  , where y0 = V0(αA + μA)/[λ(μA + 1)]. Figure 5 displays the excluded regions where the tidal forces break the dimer, which are far from the equilibrium configuration. In summary, a concentration field can direct dimers to move along the valley for a wide range of parameters, allowing to use ad hoc potentials to guide colloidal molecules.
, where y0 = V0(αA + μA)/[λ(μA + 1)]. Figure 5 displays the excluded regions where the tidal forces break the dimer, which are far from the equilibrium configuration. In summary, a concentration field can direct dimers to move along the valley for a wide range of parameters, allowing to use ad hoc potentials to guide colloidal molecules.

Figure 5. Phase portrait in the  –θ space for a dimer moving in a parabolic concentration field along cext(x, y) = λ y2/2, where θ is the dimer director measured with respect to the x axis. Here, αA − μA = 1.0 and
–θ space for a dimer moving in a parabolic concentration field along cext(x, y) = λ y2/2, where θ is the dimer director measured with respect to the x axis. Here, αA − μA = 1.0 and  . The two stable fixed points (y = 0 and θ = 0, π) indicated by black solid dots, correspond to the dimer moving along the valley of the concentration field in the positive of negative direction. In the red dashed regions, the tidal forces are strong enough to destroy the dimer.
. The two stable fixed points (y = 0 and θ = 0, π) indicated by black solid dots, correspond to the dimer moving along the valley of the concentration field in the positive of negative direction. In the red dashed regions, the tidal forces are strong enough to destroy the dimer.
Download figure:
Standard image High-resolution imageThe analysis was performed for a straight valley, but it is possible to use other geometries to guide different colloidal molecules too. Figure 6 shows the trajectory of a trimer in a circular potential cext = λ(y2 − R2)2, which presents oscillations before reaching the final circular trajectory. Due to the symmetry of the potential the motion could be either clockwise or counter-clockwise, depending on the initial condition. This circular trap also works for dimers in a qualitatively similar way.

Figure 6. Trajectory of a trimer in the x–y plane, when exposed to a circular parabolic potential cext = λ(r2 − R2)2, with λ = 10−6 and R = 50, simulated for 2000 time units using αA = 2.1, μA = 1.1 and η = 0.0125. Circles are drawn to help visualization, for radii every two distance units.
Download figure:
Standard image High-resolution image4.2. Interaction with walls
We have also implemented collisions with walls as a way of guiding the molecules. This has been previously demonstrated for Janus particles [21] but since our molecules are self-assembled and there is no hard forces keeping them together or resisting deformation, it remains to be verified that they will remain a unit when hitting a wall. We implement the wall as a linear restitutive force pointing normal to the surface The hydrodynamic interaction with walls, which despite being long range is far smaller than the elastic force [21], was not included for simplicity. However, our integration method is robust enough to add different long range fields, be it hydrodynamic or phoretic if one would like to add it. Dimers remain stable on collisions and can be guided by straight walls. Eventually, noise can detach dimers from the walls (figure 7-left). For sinusoidal walls the dimer is guided in the convex section and departs when the wall changes curvatures (figure 7-center). Trimers on the other hand, 'stick' to the wall for a moment and then bounce back (figure 7-right).

Figure 7. Trajectories of a dimer colliding with a flat and sinusoidal wall (left, center) and a trimer with a flat wall (right). The simulation parameters are αA = 2.175, μA = 1.1 and η = 0.001 25. The amplitude and wavelength of the sinusoidal wall are A = 1 and λ = 10.5, respectively.
Download figure:
Standard image High-resolution image5. Assembly of self-oscillating chains: toward colloidal molecular factories
Having demonstrated that we can produce trimers and dimers with a high yield, in a very localized way, for a wide range of parameters, and that these small colloidal molecules can be guided with walls and external potentials without destroying them, we will study a reaction path that produces the colloidal chains with dynamic function we have previously studied [19].
All the colloidal chains we reported in [19] consist of an integer number of trimers. Thus, each of these structures could be formed by the collision of trimers directed to a reaction zone by the means described in the previous sections. Colloidal molecules have many isomers, and the stability of those depends on the particular values for the phoretic charges [18, 19, 27]. For each colloidal chain there is a particular region of the phase space, where it can be stable, unstable, oscillatory or rotating. Thus, a reaction path may be stable in a region of the parameter space but not in another. We consequently restrict ourselves to study the reaction paths in the vicinity of the parameter where the oscillator is stable.
We focus on the chain A4B8 which was shown to be the smallest stable chain [19]. It can be produced by the collision of four trimers, which are disposed symmetrically in a circle of radius R = 12, with their director vectors pointing inwards. To study the robustness of the method and sample the possible outcomes of the reaction, fluctuations are added in the initial directors of the trimers (±0.1 rad) and their starting positions (±0.1) as a way to quantify the degree of precision needed experimentally to produce the chains. Also, a finite noise amplitude of η = 0.0125 is considered. Some of the possible resulting isomers are shown in figure 8.

Figure 8. Examples of A particles' trajectories for different isomers, classified according to the value of s. (a) Run-and-tumble molecule, s = 0.451; (b) rotator 1, s = 1.502; (c) rotator 2, s = 1.54; (d) oscillator, s = 1.71; and (e) broken molecule, s > 2.0.
Download figure:
Standard image High-resolution imageTo pre-classify the different isomers automatically we look at the spatial distribution of the particles' positions by mean of the time-averaged radius of gyration:
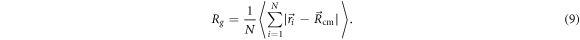
The radius of gyration is able to distinguish between the different isomers in a very appropriate manner. When varying αA, we have found that different intervals in s, give different qualitative isomers. The interval 1.71 < Rg < 1.73 selects only oscillators with an error of approximately 20%. Values of Rg > 2.0 signals the breaking of the molecules in at least two disconnected clusters with 100% accuracy. These can be dimers, trimers, or different combinations of those.
The interval Rg < 1.45 comprises different isomers that are variations of run-and-tumble molecules. The interval 1.50 < Rg < 1.54 selects two kinds of rotators that appear when varying the initial position of one of the trimers (see figure 8 for examples). After the automatic pre-classification using s, false positives and negatives are re-classified observing the trajectory of the A backbone particles, as shown in figure 8. It must be noted that due to the thermal noise present in the system there is an intrinsic noise associated to measuring Rg. Long measuring times are not always feasible since for some values of αA the molecules change between different isomers in the long term. To avoid this, we take an observation window long enough to have a few cycles (∼15) of the oscillating trajectories but short enough to not see many transition between isomers. Furthermore, some trajectories are not clearly one or other isomer, and are classified as 'others'. These trajectories though represent less than 5% of the total analyzed.
Figure 9 (top) shows the percentage of oscillatory and other structures found varying the initial configuration, obtained by radially displacing a distance δ the initial position of one of the trimers (δ < 0 meaning the trimers starts closer to the collision point). We can see that for 0.8 ≲ δ ≲ 1.8 the production rate increases by almost a factor two compared to δ = 0, resulting in that 60% of the outcomes are stable oscillators. The bottom panel of the same figure presents the effect of varying the phoretic charge αA. There is a clear maximum in the production of oscillators at α = 2.16. These results hint towards a possible general optimization of the collision's parameters to form one isomer and not others, hence the creation of complex micro structures with active colloids.
An interesting molecule is the newly found run-and-tumble isomer. This molecule has left-right symmetry and it is elongated in the direction of movement. In its head it has a trimer that tends to move faster than the rest of the the body making the structure instable. However, the head cannot scape, but only move to one side or another forcing a rearrangement of the molecule and a consequent change in the direction of motion. As an example we show different trajectories for the αA = 2.05 run-and-tumble isomer, in which the angle of turn is almost exactly 60°, see figure 10. Note how the period for the change of direction is quite constant along the trajectory, sign of a very robust feature.

Figure 9. Percentage of isomers produced in collisions of four trimers. For each point, 100 collisions were performed and the factions of produced isomers are counted (symbols). The solid lines are obtained by smoothing the results. Oscillators in blue, rotators in orange, run-and-tumblers in green, and broken molecules in red. Top: effect of changing the initial configuration varying radial displacement of one trimer, δ, while αA = 2.15 and μA = 1.1 are fixed. Bottom: effect of varying the phoretic charge αA, with δ = 1.5 and μ = 1.1 fixed.
Download figure:
Standard image High-resolution image
Figure 10. Three representative center of mass trajectories for the run-and-tumble isomer shown in figure 8(a). Here αA = 2.05 and μA = 1.1, simulated for ∼5000t0. The angle of turn depends on the chemistry of the colloids.
Download figure:
Standard image High-resolution imageFinally, we note that for these large molecules to form it is necessary that the composing trimers are soft enough to rearrange into different structures. An example of this is shown in figure 11. Particles are colored by the trimer to which they belonged initially. It is possible to see that three of the four trimers are completely destroyed and the colloids are distributed along the new molecule.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11. Collision of four trimers that produces an oscillator for αA = 2.175, μA = 1.1, and η = 0.012 5. Particles are colored according to the trimer they belonged initially and the lines indicate particles are closer than 1.25. Only the green cluster keeps its integrity after the collision, whereas all the others exchange particles.
Download figure:
Standard image High-resolution image{kind=link}
6. Discussion
We have presented, by means of simulations, a proof of concept for the directed assembly of colloidal molecules with dynamic function. In particular, we have shown how to produce, starting from monomers, the chains that have been previously reported to produce sustained beating and could function as cilia or flagella [19]. Our strategy consists of the use of external fields with particular geometries to produce highly localized dimers and trimers starting from monomers. The assembled molecules present self-motion and can be directed by means of additional external fields and walls. We have used these molecules as building blocks for more complex structures that present dynamic function. We have shown that they have stable outcomes in a certain region of design parameters and thermal noise.
In experimental terms, our production strategy of formation via collisions could be implemented with modular miscroswimmers [25, 26, 33]. The structures that could be created with those colloids however, will be different to the ones presented here, since the mobilities of the colloids are different, in particular their signs. For our method to be able to produce flagella and cilia-like molecules, one would need to experimentally modify the head-tail symmetry of the chains. This could be done by, for example, joining the two colloids of the head with lasers or creating a gradient of chemical composition along the length of the molecule. The first method has been already implemented experimentally [8].
It is worth noting that our method works for the simplest case for non-polar active colloids, namely only two different kinds of particles. More complex interactions such as anisotropic potentials, DNA patches, different chemistry of the monomers, or time-dependent external fields, could help create increasingly complex machinery following the same assisted assemble mechanism. Our results show how a vast ensemble of complex structures can emerge easily from relatively simple components in colloidal systems with an intuitive production strategy, a factory where the components and the machines are one and the same.
Acknowledgments
This research was supported by the Fondecyt Grants No. 1180791 and 3160481, and the Millennium Nucleus 'Physics of active matter' of the Millennium Scientific Initiative of the Ministry of Economy, Development and Tourism (Chile).
{kind=link}
{kind=link}